 Przejdź do treści
Przejdź do treściNazwa choroby (Dystrofia Mięśniowa Duchenne’a) pochodzi od nazwiska francuskiego neurologa Guillaume’a Benjamina-Amanda Duchenne’a, który jako pierwszy opisał to jako „paraliż przerostowy” w 1868 roku.
Duchenne de Boulogne, w całości Guillaume-Benjamin-Amand Duchenne de Boulogne (ur. 17 września 1806 w Boulogne we Francji – zm. 15 września 1875 w Paryżu.
Francuski neurolog, który jako pierwszy opisał kilka zaburzeń nerwowych i mięśniowych oraz prowadził ich leczenie. Stworzył elektrodiagnostykę i elektroterapię.
Duchenne prowadził, przez całe swoje życie, prywatną praktykę najpierw w Boulogne (1831–42), a następnie w Paryżu (1842–75). Badał wpływ stymulacji elektrycznej na chore nerwy i mięśnie.
Przedstawił pierwsze opisy kilku rodzajów zaniku mięśni i porażenia spowodowanego zaburzeniami nerwowymi, w tym:
- w 1858 Wiądu Rdzenia (łac. tabes dorsalis) – najczęstsza postać kiły trzeciorzędowej kiły układu nerwowego. Często objawem tabes dorsalis jest ataksja lokomotoryczna charakteryzująca się brakiem możliwości precyzyjnego kontrolowania ruchów własnego ciała,
- zanik mięśni spowodowany zwyrodnieniem grzbietowych kolumn rdzenia kręgowego i pni nerwów czuciowych.
W latach sześćdziesiątych XIX wieku opisał poważne postępujące osłabienie mięśni u 13 młodych chłopców. Chorobę tą jego cześć nazwano później Dystrofią Mięśniową Duchenne’a (Duchenne Muscular Dystrophy).
Jako pierwszy zapoczątkował praktykę diagnostyczną zwaną biopsją. Skonstruował do tego celu instrument (znany obecnie jako trokar Duchenne’a) do pobierania niewielkich fragmentów tkanki mięśniowej z wętrza mięśnia.

Rys. 1. Guillaume Benjamin Amand Duchenne (1806-1875)
Źródło: 1. https://fineartamerica.com/featured/guillaume-benjamin-amand-duchenne-de-mary-evans-picture-library.html
2. https://www.britannica.com/biography/Duchenne-de-Boulogne
Dystrofia Mięśniowa Duchenne’a (DMD):
DMD jest ciężką, postępującą chorobą wyniszczającą mięśnie. Prowadzi ona do trudności w poruszaniu się, a ostatecznie do konieczności wspomagania oddychania oraz innych chorób współistniejących i przedwczesnej śmierci.
Przyczyną choroby są mutacje w genie dystrofiny, największym ludzkim genie zlokalizowanym w locus Xp21.2. W przypadku DMD mutacja ta skutkuje poważnym brakiem dystrofiny (<5%);
Dystrofina pełni funkcje strukturalne, chroniąc komórkę mięśniową przed napięciem w czasie skurczu i rozkurczu włókna mięśniowego. Brak dystrofiny powoduje upośledzenie mięśni szkieletowych, mięśnia sercowego (prowadząc do kardiomiopatii) i mięśni gładkich np w jelitach. Powoduje, że zanika motoryczna składowa aktywności człowieka.

Rys. 1. Schemat dystrofiny łączącej sieć aktynową z transbłonowymi składnikami DGC*
Źródło: https://www.bioprocessonline.com/doc/simple-western-advances-cutting-edge-duchenne-muscular-dystrophy-research-0001
*DGC – Dystrophin-associated protein complex (Kompleks białkowy związany z dystrofiną)
Ponieważ DMD dziedziczy się w sposób recesywny sprzężony z chromosomem X (należy do dystrofii XLR X-Linked Recessive – Recesywnie Sprzężony z chromosomem X). Dotyka głównie chłopców, którzy otrzymali zmutowany chromosom X od bezobjawowej chorej matki lub matki z łagodnym przebiegiem choroby, która jest tzw. nosicielką. W przypadku urodzenia córek, dziewczynki te będą nosicielkami wadliwego genu i przekażą go swoim dzieciom płci męskiej. Choroba objawia się więc głównie przez dziedziczenie. Jednak nie tylko dziedziczenie jest powodem choroby. Może nią być także mutacja spontaniczna. Kliknij aby poczytać o mutacjach.
W przypadku mutacji spontanicznej wada genetyczna pojawia się w jajeczku kobiety, które po połączeniu się z plemnikiem tworzy zygotę. Komórka zygoty ulega dalszym podziałom, a komórki postałe w wyniku tych podziałów dziedziczą wadę genetyczną, którą posiadło jajeczko kobiety.
Prawdopodobieństwo zachorowania wynosi 50%. Oznacza to, że urodzić się może zarówno chory/a jak i zdrowy/a chłopiec/dziewczynka w zależności od tego, który chromosom otrzyma od matki. Jeżeli otrzyma wadliwy będzie chory/a jeżeli prawidłowy będzie zdrowy/a.
Należy tu zauważyć, że choroba może zaniknąć w rodzinie i nie dotyczyć kolejnych pokoleń jeżeli dzieci otrzymają prawidłowy chromosom od matki.
Prawdopodobieństwo, że chory chłopiec będzie miał zdrowych synów wynosi 100%. Jest też 100% prawdopodobieństwo, że jego córki będą nosicielkami.
Badania prowadzone w ostatnich latach znacznie pogłębiły wiedzę na temat pierwotnych i wtórnych mechanizmów patogenetycznych tej choroby.
Ustalono wytyczne dotyczące multidyscyplinarnej opieki nad chorymi z Dystrofią Mięśniową Duchenne’a, w tym uzyskania diagnozy genetycznej i zarządzania różnymi aspektami choroby.
Wiele nowych potencjalnych terapii jest w fazie badań klinicznych. Spośród nich dużo jest już takich, które organy regulacyjne takie jak EMA w UE i FDA w USA zatwierdziły warunkowo. Celem wielu z tych terapii jest przywrócenie brakującego białka dystrofiny lub leczenie chorób/patologii wtórnych.
Objawy DMD pojawiające się przed szóstym rokiem życia to:
- postępujące osłabienie mięśni i ich atrofia,
- hipertrofia (przerost) mięśni łydki,
- zmęczenie,
- chodzenie na palcach,
- trudności we wchodzeniu i schodzeniu po schodach,
- częste upadki,
- opóźnienie w rozwoju motorycznym,
- problemy z oddychaniem,
- problemy z uczeniem się, w tym możliwe problemy z pamięcią krótkotrwałą,
- opóźnienie w rozwoju werbalnym, w tym z rozpoczęciem mówienia i dalszym rozwojem umiejętności językowych,
- skolioza,
- trudności z siadaniem, staniem i chodzeniem,
- kaczkowaty/kołyszący się chód,
- trudności w bieganiu.
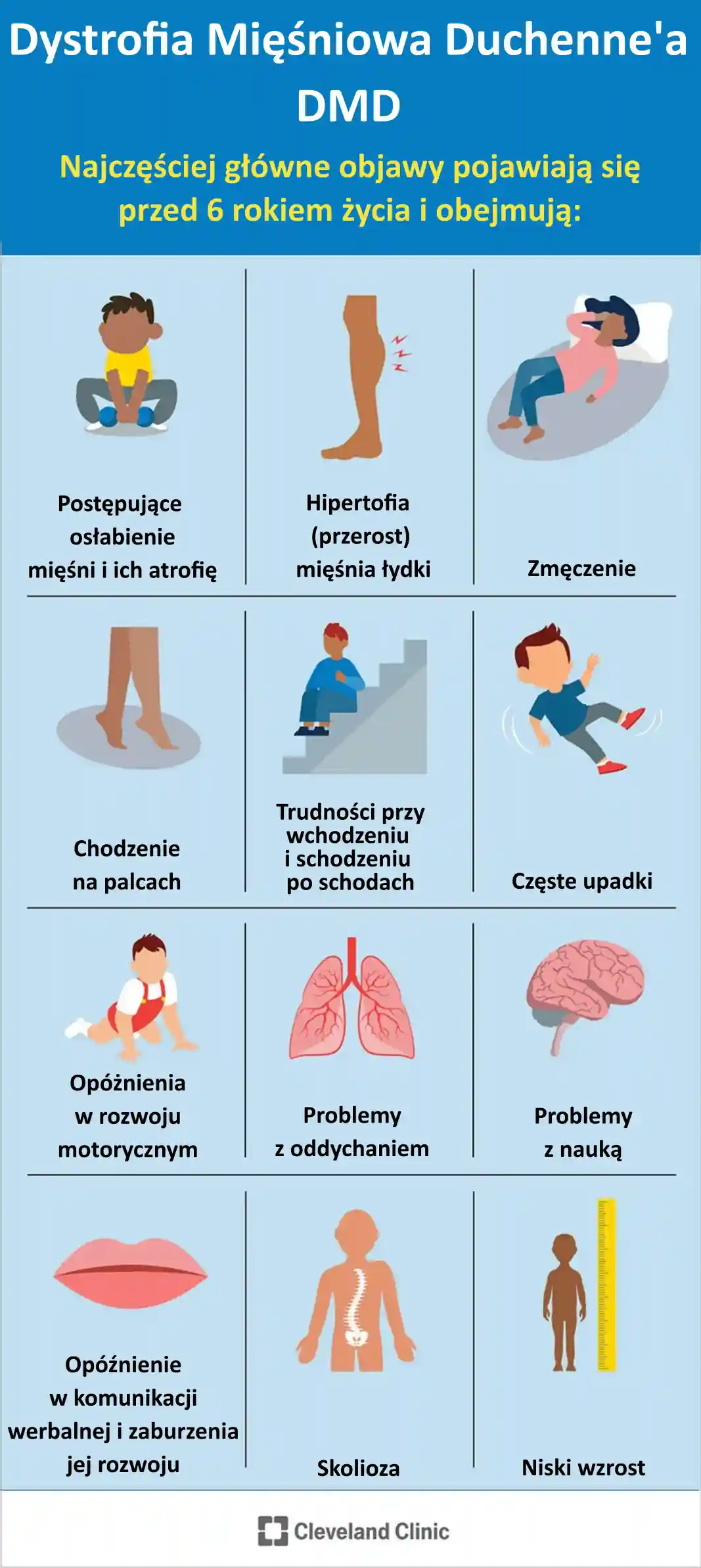
Rys. 2. Najczęstrze objawy DMD pojawiające się przed 6 rokiem życia
Źródło: https://my.clevelandclinic.org/health/diseases/23538-duchenne-muscular-dystrophy-dmd
W 1879 roku neurolog Sir William Richard Gowers opisał najbardziej znaczący i charakterystyczny objaw u pacjentów z Dystrofią Mięśniową Duchenne’a. Polega on na „wspinaniu się” po udach za pomocą rąk, w celu przezwyciężenia słabości miednicy i mięśni nóg. Objaw od nazwiska naukowca nazwano “Objawem Gowers’a” (Gowers sign).