
RECENZJA | TOM 17, NUMER 3, P251-267,01 MARCA 2018 R.
Diagnostyka i postępowanie w DMD - część 1
Diagnostyka i postępowanie w dystrofii mięśniowej Duchenne’a, część 1:
diagnostyka oraz postępowanie nerwowo-mięśniowe, rehabilitacyjne, endokrynologiczne, żołądkowo-jelitowe i żywieniowe
Spis treści________________
Add a header to begin generating the table of contents
Streszczenie
Od czasu publikacji rozważań dotyczących opieki nad dystrofią mięśniową Duchenne’a (DMD) w 2010 r. rozwinęła się interdyscyplinarna opieka nad tą ciężką, postępującą chorobą nerwowo-mięśniową. W połączeniu z poprawą przeżywalności pacjentów nastąpiła zmiana w kierunku bardziej przewidujących strategii diagnostycznych i terapeutycznych, z ponownym skupieniem się na jakości życia pacjentów. W 2014 r. powołano komitet sterujący złożony z ekspertów z wielu różnych dyscyplin w celu zaktualizowania rozważań dotyczących opieki nad DMD z 2010 r., mając na celu poprawę opieki nad pacjentem. Nowe rozważania dotyczące opieki mają na celu zaspokojenie potrzeb pacjentów o wydłużonym przeżyciu, dostarczenie wskazówek dotyczących postępów w ocenie i interwencjach oraz rozważenie implikacji pojawiających się genetycznych i molekularnych terapii dla DMD. Komitet zidentyfikował 11 tematów, które należy uwzględnić w aktualizacji, osiem z nich zostało omówionych w pierwotnych rozważaniach dotyczących opieki. Trzy nowe tematy to opieka podstawowa i postępowanie w nagłych wypadkach, leczenie hormonalne oraz zmiany w opiece przez całe życie. W części 1 tego trzyczęściowego uaktualnienia przedstawiamy rozważania dotyczące opieki nad rozpoznaniem DMD oraz leczeniem nerwowo-mięśniowym, rehabilitacyjnym, endokrynologicznym (wzrost, dojrzewanie i niewydolność nadnerczy) oraz żołądkowo-jelitowym (w tym odżywianiem i dysfagią). żołądkowo-jelitowe i żywieniowe
Wstęp
Dystrofia Mięśniowa Duchenne’a (DMD) jest śmiertelnym, sprzężonym z chromosomem X, recesywnym zaburzeniem nerwowo-mięśniowym spowodowanym mutacjami w genie dystrofiny, które skutkują brakiem lub niewystarczającą dystrofiną funkcjonalną, białkiem cytoszkieletu, które zapewnia siłę, stabilność i funkcjonalność włókien mięśniowych. Częstość występowania DMD została zgłoszona jako 15,9 przypadków na 100 000 żywych urodzeń mężczyzn w USA i 19,5 przypadków na 100 000 żywych urodzeń mężczyzn w Wielkiej Brytanii.1,2,3
Postępujące uszkodzenie i zwyrodnienie mięśni występuje u osób z DMD, powodując osłabienie mięśni, związane z tym opóźnienia motoryczne, utratę zdolności poruszania się, zaburzenia oddychania i kardiomiopatię. Chociaż przebieg kliniczny zajęcia mięśni szkieletowych i serca może być różny, śmierć zwykle następuje w wyniku upośledzenia czynności serca lub układu oddechowego.4,5 To jest część 1 trzyczęściowej aktualizacji rozważań dotyczących opieki nad DMD z 2010 roku,6,7,8 które zostało wsparte przez amerykańskie Centrum Kontroli i Prewencji Chorób (CDC) z udziałem sieci TREAT-NMD zajmującej się chorobami nerwowo-mięśniowymi, Stowarzyszenie Dystrofii Mięśniowej i Dystrofia Mięśniowa Projektu Rodzicielskiego.
Decyzja o aktualizacji rozważań dotyczących opieki była podyktowana kilkoma ważnymi wydarzeniami. Po pierwsze, dzięki opiece wielodyscyplinarnej, przeżywalność pacjentów z DMD uległa poprawie, a podejście diagnostyczne i terapeutyczne odpowiednich podspecjalizacji ewoluuje.9,10,11,12 Wraz z coraz szerszym uświadomieniem sobie przedłużonego przeżycia, wiele podspecjalizacji przesunęło się na bardziej przewidujące strategie diagnostyczne i terapeutyczne, aby osiągnąć profilaktykę, wczesną identyfikację i leczenie przewidywalnych i potencjalnie modyfikowalnych powikłań choroby. Po drugie, oczekiwaniu dłuższego przeżycia towarzyszy coraz większy nacisk na jakość życia i zarządzanie psychospołeczne. Ponadto istnieje pilna potrzeba koordynowania i usprawniania przejścia pacjentów z dzieciństwa do dorosłości. Po trzecie, ta aktualizacja była podyktowana rosnącym doświadczeniem z istniejącymi terapiami oraz oczekiwaniem pojawiających się genetycznych i molekularnych terapii DMD.13 W szczególności dostępne są nowe informacje na temat skuteczności, skutków ubocznych i ograniczeń glikokortykoidów,14,15
Aby ocenić nowe terapie, należy zidentyfikować klinicznie znaczące i wiarygodne biomarkery i miary wyników.
W części 1 niniejszego Przeglądu omówimy następujące tematy: diagnostyka, postępowanie nerwowo-mięśniowe, postępowanie rehabilitacyjne, postępowanie endokrynologiczne (w tym wzrost, dojrzewanie i niewydolność nadnerczy) oraz postępowanie żołądkowo-jelitowe (w tym odżywianie i dysfagia). Części 2 i 3 tego przeglądu opisują kwestie związane z opieką w innych obszarach tematycznych, w tym rozszerzoną sekcję dotyczącą zarządzania psychospołecznego oraz nowe sekcje dotyczące podstawowej opieki zdrowotnej, postępowania w sytuacjach nagłych oraz zmiany opieki przez całe życie. Rysunek1 przedstawia przegląd ocen i interwencji we wszystkich tematach, uporządkowanych według stadium choroby.

Rysunek 1:
Kompleksowa opieka nad osobami z mięśniami Duchenne’a dystrofia Opiekę nad pacjentami z dystrofią ięśniową Duchenne’a zapewnia a multidyscyplinarny zespół pracowników służby zdrowia; specjalista nerwowo-mięśniowy pełni funkcję głównego klinicysty. Rysunek obejmuje oceny i interwencje we wszystkich stadiach choroby i tematach omówionych w tym trzyczęściowym Przeglądzie.
*Echokardiogram dla pacjentów w wieku 6 lat lub młodszych. MRI serca u pacjentów starszych niż 6 lat.
Metody
W 2014 roku, w oparciu o swoje perspektywy kliniczne i wiedzę specjalistyczną, komitet sterujący Grupy Roboczej ds. Opieki nad DMD (CCWG) zidentyfikował 11 tematów, które należy uwzględnić w tej aktualizacji rozważań dotyczących opieki nad DMD z 2010 roku.6
Osiem tematów zostało poruszone w pierwotnych rozważaniach dotyczących opieki: (1) diagnoza, (2) postępowanie nerwowo-mięśniowe, (3) postępowanie rehabilitacyjne, (4) postępowanie żołądkowo-jelitowe i odżywianie, (5) postępowanie oddechowe, (6) postępowanie kardiologiczne, ( 7) postępowanie ortopedyczne i chirurgiczne oraz (8) postępowanie psychospołeczne. Nowe są trzy tematy: (9) opieka podstawowa i postępowanie w nagłych wypadkach, (10) leczenie endokrynologiczne (w tym wzrost, dojrzewanie, niedoczynność kory nadnerczy i zdrowie kości) oraz (11) zmiana opieki przez całe życie.
Wskazówki zawarte w tej aktualizacji nie są zwykle oparte na dowodach. Jak to jest typowe dla rzadkiej choroby, przeprowadzono kilka dużych randomizowanych badań kontrolowanych (RCT) dotyczących DMD, z wyjątkiem badań nad kortykosteroidami.16 Dlatego też, jeśli chodzi o względy opieki nad DMD z 2010 roku,6,7 wytyczne zostały opracowane przy użyciu metody, która pyta grupę ekspertów o stosowność i konieczność określonych ocen i interwencji, przy użyciu scenariuszy klinicznych.17 Metoda ta ma na celu zobiektywizowanie opinii ekspertów i uczynienie wytycznych prawdziwym odzwierciedleniem poglądów i praktyk panelu ekspertów, w oparciu o ich interpretację i zastosowanie istniejącej literatury naukowej. Stosując to podejście, byliśmy w stanie stworzyć niezbędny zestaw narzędzi do opieki nad DMD; Zalecane są tylko oceny i interwencje, które zostały uznane za odpowiednie i konieczne.
Dokonano kompleksowego przeglądu literatury w celu zidentyfikowania artykułów związanych z opieką DMD dla każdego obszaru tematycznego, z dodaniem słów kluczowych dla nowych tematów. Pełny opis strategii przeglądu literatury, tabela wyszukiwanych terminów oraz streszczenia odpowiedniej literatury są dostępne w załączniku . Na podstawie wyników wyszukiwania komitet sterujący wybrał artykuły zawierające informacje, które mogą wymagać zaktualizowania rozważań dotyczących opieki na rok 2010. Scenariusze kliniczne zostały następnie opracowane na podstawie treści tych artykułów. Dla każdego z 11 obszarów tematycznych powołano komisję ekspertów. Korzystając z metody adekwatności (RAM), RAND Corporation–University of California Los Angeles Appropriateness Method (RAM),6,17 komisje ustaliły, które oceny i interwencje są zarówno odpowiednie, jak i konieczne dla różnych scenariuszy klinicznych. W przypadku procesu RAM komisje miały dwie rundy w celu ustalenia stosowności, po których następowała jedna lub dwie rundy w razie konieczności. W poniższych sekcjach nie były wymagane wszystkie etapy dwustopniowego procesu oceny pamięci RAM, albo z powodu braku nowej literatury od czasu opracowania rozważań dotyczących opieki w 2010 r., albo dlatego, że natychmiast osiągnięto jednomyślne porozumienie między członkami komitetu co do stosowności i konieczności interwencji: diagnostyka, postępowanie nerwowo-mięśniowe, postępowanie oddechowe, postępowanie kardiologiczne, postępowanie ortopedyczne i chirurgiczne oraz postępowanie psychospołeczne. Dodatkowo, metoda RAM nie została uznana za odpowiednią dla dwóch nowych sekcji: podstawowej opieki zdrowotnej i zarządzania w nagłych wypadkach, i przejścia opieki przez całe życie. Komisje zajmujące się tymi sekcjami osiągnęły konsensus podczas dyskusji bez pierwszej oceny scenariuszy klinicznych.
Diagnoza
Osiągnięcie terminowej i dokładnej diagnozy DMD jest kluczowym aspektem opieki. Metoda diagnozowania DMD nie zmieniła się znacząco od 2010 roku ( Rysunek 2 ).6
Proces diagnostyczny zazwyczaj rozpoczyna się we wczesnym dzieciństwie po zauważeniu sugestywnych oznak i objawów, takich jak osłabienie, niezdarność, objaw Gowersa, trudności ze wchodzeniem po schodach lub chodzeniem na palcach. Szybkie skierowanie do specjalisty nerwowo-mięśniowego, z udziałem genetyka lub doradcy genetycznego, może uniknąć opóźnienia w diagnostyce.18
Rzadziej diagnoza jest uważana za wynik opóźnienia rozwojowego19 lub zwiększone stężenia enzymów surowicy, takich jak aminotransferaza alaninowa, aminotransferaza asparaginianowa, dehydrogenaza mleczanowa lub kinaza kreatynowa. Niekiedy zwiększone stężenie aminotransferazy alaninowej, asparaginianowej lub dehydrogenazy mleczanowej powoduje niewłaściwe skupienie się na zaburzeniach czynności wątroby, opóźniając rozpoznanie DMD.

Rysunek 2: Opisane wczesne oznaki i objawy DMD są oparte na Ciafaloni i współpracownikach.18 DMD=Dystrofia mięśniowa Duchenne’a.
Ponieważ około 70% osób z DMD ma delecję lub duplikację jednego lub wielu eksonów w genie dystrofiny, badanie delecji i duplikacji genu dystrofiny jest zwykle pierwszym testem potwierdzającym. Testowanie najlepiej przeprowadzać za pomocą multipleksowej amplifikacji sondy zależnej od ligacji (MLPA)20 lub porównawcza macierz hybrydyzacji genomowej,21 ponieważ użycie multipleksowego PCR może jedynie zidentyfikować delecje. Identyfikacja granic mutacji delecji lub duplikacji przez MLPA lub macierz porównawczej hybrydyzacji genomowej może wskazywać, czy przewiduje się, że mutacja zachowa lub zakłóci ramkę odczytu. Jeśli wynik testu delecji lub duplikacji jest negatywny, należy przeprowadzić sekwencjonowanie genetyczne w celu zbadania pozostałych typów mutacji przypisywanych DMD (około 25–30%).22
Mutacje te obejmują mutacje punktowe (nonsens lub missens), małe delecje i małe duplikacje lub insercje, które można zidentyfikować za pomocą sekwencjonowania nowej generacji.23,24,25
Wreszcie, jeśli testy genetyczne nie potwierdzą klinicznej diagnozy DMD, próbka z biopsji mięśnia powinna zostać przebadana na obecność białka dystrofiny za pomocą immunohistochemii kriosekcji tkanek lub metodą western blot ekstraktu białka mięśniowego.
Nosicielki
Członkowie rodziny osoby z DMD powinni otrzymać poradę genetyczną w celu ustalenia, kto jest zagrożony nosicielstwem. Badanie na nosicielstwo jest zalecane u krewnych chłopca lub mężczyzny, u których potwierdzono genetycznie DMD. Jeśli krewny jest dzieckiem, należy przestrzegać wytycznych etycznych Amerykańskiego Towarzystwa Medycznego dotyczących badań genetycznych dzieci.26
Po zidentyfikowaniu nosiciele płci żeńskiej mają do rozważenia kilka opcji reprodukcyjnych, w tym preimplantacyjną diagnostykę genetyczną lub prenatalne testy genetyczne poprzez pobieranie próbek kosmówki lub płynu owodniowego. Kobiety nosicielki wymagają również oceny lekarskiej i obserwacji, jak opisano w części dotyczącej postępowania kardiologicznego w części 2 niniejszego Przeglądu.
Badanie przesiewowe noworodka
Możliwość przeprowadzenia badań przesiewowych noworodków w kierunku DMD została po raz pierwszy pokazana w połowie lat 70.27 poprzez pomiar stężeń kinazy kreatynowej z wysuszonych plamek krwi. Ostatnio pojawił się dwupoziomowy system diagnostyki przesiewowej noworodków,2 w których próbki, które wykazały zwiększone stężenie kinazy kreatynowej, były następnie testowane pod kątem mutacji genu dystrofiny. Badania przesiewowe noworodków w kierunku DMD przeprowadzono w kilku krajach, ale większość z nich została przerwana,2,28 a DMD nie jest obecnie uwzględniony w zalecanym panelu jednolitego badania przesiewowego,29 co jest w dużej mierze ograniczone do zaburzeń o początku noworodkowym, w przypadku których wczesne leczenie daje lepsze wyniki. Jednak ponowne zainteresowanie badaniami przesiewowymi noworodków wzrosło dzięki wsparciu ze strony zainteresowanych stron oraz ponieważ pojawiające się terapie DMD mogą okazać się najskuteczniejsze, jeśli zostaną rozpoczęte przed wystąpieniem objawów.30,31
Postępowanie neurologiczne
Po postawieniu diagnozy lekarz neurolog będzie pełnił funkcję głównego klinicysty, biorąc na siebie całkowitą odpowiedzialność za opiekę nad osobą z DMD oraz pełniąc wiele ról i obowiązków przez całe życie tej osoby (panel1). Lekarz neurolog ma wyjątkowe kwalifikacje do prowadzenia pacjentów i ich rodzin przez coraz bardziej złożony i technologiczny krajobraz diagnostyczny i terapeutyczny współczesnej opieki nad DMD.
Role i obowiązki lekarza neurologa w opiece nad pacjentami z Dystrofią Mięśniową Duchenne’a
- Ocena i charakterystyka, unikalnego dla każdego pacjenta, przebiegu choroby w czasie za pomocą zwalidowanych narzędzi oceny, w celu ustalenia oczekiwanego przebiegu klinicznego choroby pacjenta oraz przedstawienia rokowań i potencjalnych powikłań
- Wykorzystanie informacji z oceny w celu wyboru interwencji terapeutycznych, które definiują indywidualny plan leczenia, zaprojektowany tak, aby spełniał szczególne potrzeby i cele każdego pacjenta i rodziny, optymalizując wyniki i jakość życia zdefiniowane indywidualnie przez pacjenta i rodzinę
- Zaangażowanie określonych klinicystów mogących przeprowadzić wyznaczone oceny, interwencje i plan leczenia. Najlepiej w kontekście dedykowanej, wielodyscyplinarnej kliniki DMD prowadzonej, administrowanej i koordynowanej przez specjalistę, lekarza neurologa; asystować w opiece nad nosicielkami, w tym w ocenie kardiologicznej
- Bycie pierwszym doradcą medycznym dla pacjentów i ich rodzin podczas określania i modyfikowania ich indywidualnych celów opieki wraz z rozwojem choroby w czasie, pomagając im spersonalizować analizę ryzyka w stosunku do korzyści stosowanych interwencji terapeutycznych, w tym:
- Interwencji technologicznych w leczeniu układu oddechowego i kardiologicznego
- Zabiegów chirurgicznych i niechirurgicznych, takich jak usztywnienie kręgosłupa, postępowanie z przykurczami oraz zapewnienie pomocy i urządzeń medycznych
- Interwencji farmakologicznych, takich jak terapia glikokortykosteroidami, pojawiających się nowych terapii i udziału pacjentów w badaniach klinicznych leków eksperymentalnych
- Bycie orędownikiem wysokiej jakości opieki nad pacjentami z DMD w ośrodkach zdrowia zajmujących się pacjentami oraz w społecznościach lokalnych, w których pacjenci ci żyją ,podnosząc takie kwestie, jak przejście z opieki pediatrycznej na opiekę kliniczną dla dorosłych oraz zapewnienie opieki szpitalnej, która ma na celu zajęcie się wyjątkowymi medycznymi, fizycznymi i psychospołecznymi potrzebami pacjentów
- Pomoc pacjentom i ich rodzinom w radzeniu sobie z opieką u schyłku życia w sposób, który chroni komfort, godność i jakość życia, zgodnie z potrzebami zdefiniowanymi przez każdego pacjenta i ich rodziny
Ocena kliniczna
Spójna i powtarzalna ocena kliniczna funkcji nerwowo-mięśniowej wykonana przez przeszkolonych lekarzy stanowi podstawę leczenia DMD. Oceny opisane w rozważaniach dotyczących opieki z 2010 r. pozostają aktualne, a kliniki powinny stosować zestaw testów, z którym czują się komfortowo i dla których rozumieją kliniczne korelacje. Członkowie zespołu multidyscyplinarnego muszą współpracować, aby zoptymalizować spójność i uniknąć niepotrzebnego powielania testów. Sugerowane oceny przedstawiono w załącznikui są omówione w części dotyczącej postępowania rehabilitacyjnego. Nowsze badania wykazały wartość minimalnych klinicznie istotnych różnic, możliwości predykcyjnych standaryzowanych ocen czynnościowych oraz zakresy optymalnej reaktywności, potwierdzając znaczenie standaryzowanych ocen czynnościowych przez całe życie pacjenta.32,33,34,35
Ponadto nowe narzędzia oceny pomagają w kierowaniu postępowaniem ze starszymi osobami, które nie potrafią chodzić, ilustrując znaczenie testów klinicznych przez całe życie.
Interwencje
Fizjoterapia, jak opisano w części dotyczącej postępowania rehabilitacyjnego, oraz leczenie glikokortykosteroidami pozostają podstawą leczenia DMD i powinny być kontynuowane po utracie możliwości poruszania się. Rysunek 3 opisuje inicjację i stosowanie glikokortykoidów.36
Wykazano, że korzyści z długotrwałej terapii glikokortykosteroidami obejmują utratę zdolności poruszania się w późniejszym wieku, zachowanie funkcji kończyny górnej i układu oddechowego oraz unikanie operacji skoliozy.37
Ostatnie badania potwierdzają korzyści z rozpoczęcia przyjmowania glikokortykosteroidów u młodszych dzieci przed znacznym pogorszeniem stanu fizycznego;38,39 trwające badanie ( identyfikator ClinicalTrials.gov NCT02167217 ) nad dawkowaniem w weekendy u chłopców w wieku poniżej 30 miesięcy wkrótce dostarczy dodatkowych informacji. Chociaż korzyści z terapii glikokortykosteroidami są dobrze znane, pozostaje niepewność co do tego, które glikokortykoidy są najlepsze iw jakich dawkach.40
Te niepewności zwiększają ryzyko niedostatecznego lub nadmiernego leczenia, co może zakłócić wyniki prób innowacyjnych terapii. Wielkoskalowe badania historii naturalnej i badania kohortowe potwierdzają wydłużenie okresu poruszania się ze średnio 10,0 lat u osób leczonych kortykosteroidami przez mniej niż 1 rok do średnio 11,2 lat u osób leczonych prednizonem codziennie i 13,9 lat w osoby przyjmujące codziennie deflazakort.41
W kilku badaniach dawkowanie prednizonu tylko w weekendy wykazało skuteczność równą dawkowaniu codziennemu.41,42
W badaniu klinicznym III fazy z podwójnie ślepą próbą porównano deflazakort 0,9 mg/kg dziennie, deflazakort 1,2 mg/kg dziennie, prednizon 0,75 mg/kg dziennie i placebo. Wszystkie grupy leczenia poprawiły siłę mięśni w porównaniu z placebo, a deflazakort wiązał się z mniejszym przyrostem masy ciała niż prednizon.14
Stosunek korzyści do ryzyka deflazakortu w porównaniu z prednizonem jest dalej badany w trwającej podwójnie ślepej próbie.15
Te niepewności zwiększają ryzyko niedostatecznego lub nadmiernego leczenia, co może zakłócić wyniki prób innowacyjnych terapii. Wielkoskalowe badania historii naturalnej i badania kohortowe potwierdzają wydłużenie okresu poruszania się ze średnio 10,0 lat u osób leczonych kortykosteroidami przez mniej niż 1 rok do średnio 11,2 lat u osób leczonych prednizonem codziennie i 13,9 lat w osoby przyjmujące codziennie deflazakort.41
W kilku badaniach dawkowanie prednizonu tylko w weekendy wykazało skuteczność równą dawkowaniu codziennemu.41,42
W badaniu klinicznym III fazy z podwójnie ślepą próbą porównano deflazakort 0,9 mg/kg dziennie, deflazakort 1,2 mg/kg dziennie, prednizon 0,75 mg/kg dziennie i placebo. Wszystkie grupy leczenia poprawiły siłę mięśni w porównaniu z placebo, a deflazakort wiązał się z mniejszym przyrostem masy ciała niż prednizon.14
Stosunek korzyści do ryzyka deflazakortu w porównaniu z prednizonem jest dalej badany w trwającej podwójnie ślepej próbie.15
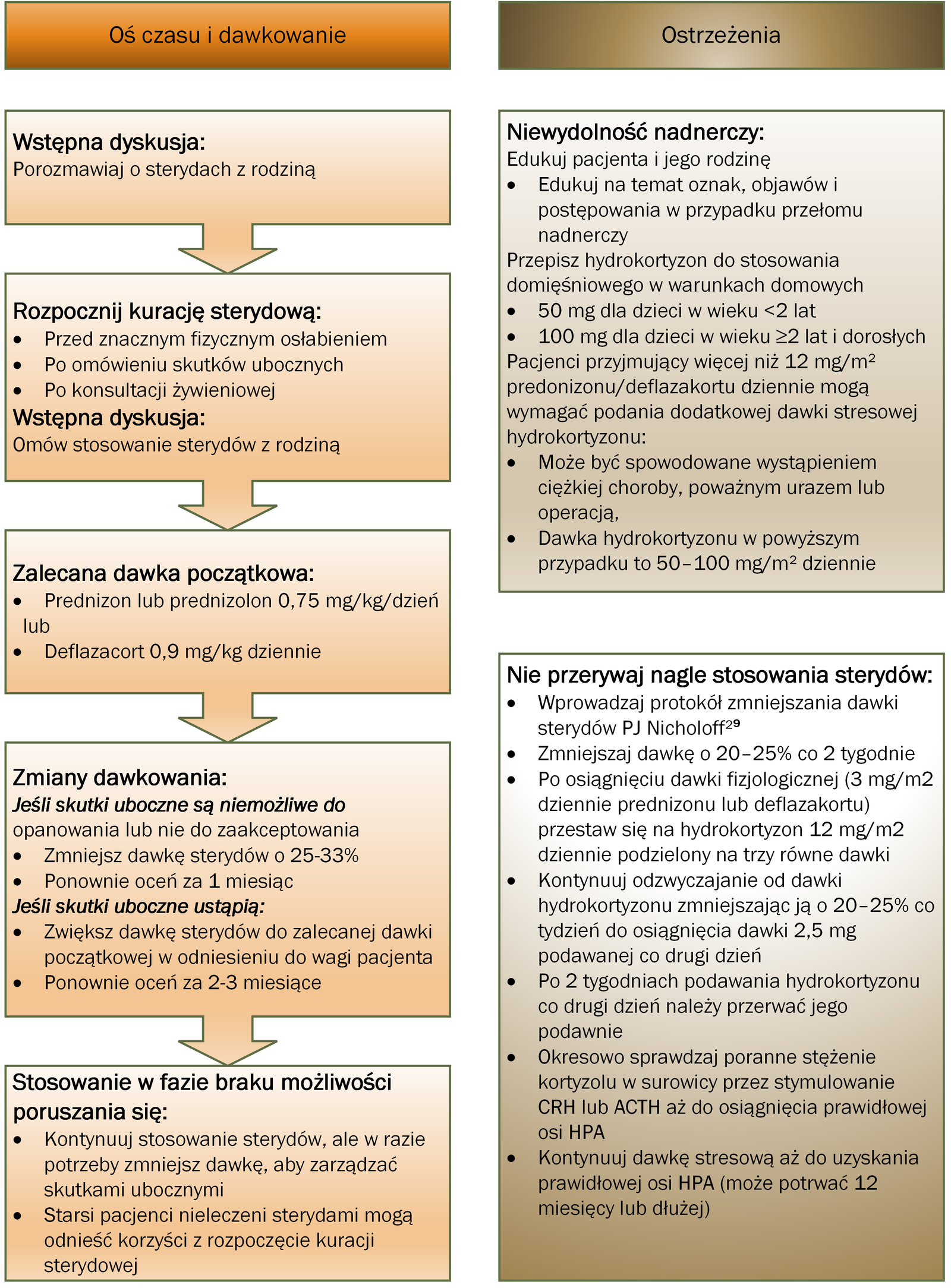
Rysunek 3:
Rozważania dotyczące opieki podczas rozpoczynania i stosowania glikokortykosteroidów (steroidów) u pacjentów z dystrofią mięśniową Duchenne’a
Nowe terapie
Plan opracowywania leków dla DMD zmienił się radykalnie od czasu publikacji rozważań dotyczących opieki w 2010 roku, a pełna lista badań dotyczących leczenia DMD stale się zmienia; Aktualne informacje są dostępne na stronie ClinicalTrials.gov i Międzynarodowej Platformie Rejestru Badań Klinicznych WHO . DMD jest rzadką chorobą, a rosnąca liczba badań nad DMD stanowi wyzwanie dla zdolności do prowadzenia badań klinicznych ze względu na małą liczbę pacjentów kwalifikujących się do udziału. Oczekuje się, że potrzeba optymalizacji rekrutacji pacjentów będzie promować inicjatywy wspierające gotowość do badań, takie jak rejestry pacjentów, identyfikacja klinicznie istotnych wskaźników wyników oraz badania historii naturalnej.
W sierpniu 2014 r. Komisja Europejska przyznała atalurenowi warunkowe pozwolenie na dopuszczenie do obrotu w Unii Europejskiej, skierowane do około 11% chłopców z DMD wywołaną kodonem stop w genie dystrofiny.43,44
We wrześniu 2016 r. Amerykańska Agencja ds. Żywności i Leków (FDA) zatwierdziła stosowanie eteplirsenu, który jest skierowany do około 13% chłopców z mutacją w genie dystrofiny, która jest podatna na pominięcie eksonu 51.45
poprzez przyspieszoną ścieżkę zatwierdzania. Ataluren i eteplirsen są pierwszymi z serii terapii specyficznych dla mutacji, które uzyskały aprobatę organów regulacyjnych. Inne terapie odbudowy dystrofiny są w trakcie opracowywania, a niektóre są bliskie lub są w trakcie przeglądu regulacyjnego.13
FDA przyznała również pełną aprobatę dla deflazakort, czyniąc go pierwszym glikokortykoidem z oznakowanym wskazaniem specjalnie dla DMD.
We wrześniu 2016 r. Amerykańska Agencja ds. Żywności i Leków (FDA) zatwierdziła stosowanie eteplirsenu, który jest skierowany do około 13% chłopców z mutacją w genie dystrofiny, która jest podatna na pominięcie eksonu 51.45
poprzez przyspieszoną ścieżkę zatwierdzania. Ataluren i eteplirsen są pierwszymi z serii terapii specyficznych dla mutacji, które uzyskały aprobatę organów regulacyjnych. Inne terapie odbudowy dystrofiny są w trakcie opracowywania, a niektóre są bliskie lub są w trakcie przeglądu regulacyjnego.13
FDA przyznała również pełną aprobatę dla deflazakort, czyniąc go pierwszym glikokortykoidem z oznakowanym wskazaniem specjalnie dla DMD.
Inne klasy leków w badaniach nad DMD obejmują leki skierowane na miostatynę, cząsteczki przeciwzapalne i przeciwutleniające, związki zmniejszające zwłóknienie, leki poprawiające rozszerzenie naczyń krwionośnych, leki poprawiające funkcję mitochondriów oraz leki regulujące utrofinę.46
Jednak bez ukończonych badań klinicznych i zatwierdzenia przez organy regulacyjne żaden z tych leków nie może być przepisany osobom z DMD.
Jednak bez ukończonych badań klinicznych i zatwierdzenia przez organy regulacyjne żaden z tych leków nie może być przepisany osobom z DMD.
Postępowanie rehabilitacyjne
DMD charakteryzuje się dobrze znanymi wzorcami postępującej degeneracji i osłabienia mięśni, kompensacji postawy, ryzyka postępującego przykurczu i deformacji oraz utraty funkcjonalnej wynikającej z niedoboru dystrofiny.6,7
Ulepszone zarządzanie DMD spowodowało wydłużenie chodu,47 zmniejszona częstość występowania ciężkich przykurczów i deformacji, w tym skoliozy,37 oraz przedłużona funkcja i uczestnictwo we wszystkich dziedzinach życia.47,48
Personel rehabilitacyjny obejmuje lekarzy, fizjoterapeutów, terapeutów zajęciowych, logopedów, ortotyków i dostawców trwałego sprzętu medycznego. Panel 2 i załącznik przedstawiają przegląd sugerowanych ocen i interwencji. Postępowanie rehabilitacyjne wymaga zrozumienia patologii DMD, patokinezjologii, historii naturalnej i progresji choroby; świadczeniodawcy powinni rozważyć cele i styl życia każdej osoby, aby zoptymalizować jakość życia przez całe życie.7
Ocena i postępowanie wyprzedzające muszą być zapewnione we wszystkich dziedzinach Międzynarodowej Klasyfikacji Funkcjonowania, Niepełnosprawności i Zdrowia (ICF) od momentu rozpoznania, aby zminimalizować przykurcze, deformacje, utratę funkcji, uszkodzoną integralność skóry, ból i obniżony stan krążeniowo-oddechowy.
Oceny i interwencje rehabilitacyjne we wszystkich stadiach choroby u pacjentów z dystrofią mięśniową Duchenne’a
Oszacowanie
Wielodyscyplinarna ocena rehabilitacji co 6 miesięcy lub częściej, jeśli występują obawy, zmiana statusu lub szczególne potrzeby ( załącznik )
Interwencja
Leczenie bezpośrednie
Leczenie bezpośrednie realizowane przez fizjoterapeutów, terapeutów zajęciowych i logopedów, dostosowane do indywidualnych potrzeb, stadium choroby, odpowiedzi na terapię i tolerancji, prowadzone przez całe życie pacjenta
Zapobieganie przykurczom i deformacjom
- Codzienne profilaktyczne rozciąganie w domu 4–6 razy w tygodniu; regularne rozciąganie kostek, kolan i bioder; rozciąganie nadgarstków, dłoni i szyi później, jeśli jest to wskazane w ocenie
- Rozciąganie w przypadku struktur, o których wiadomo, że są zagrożone przykurczem i deformacją * i te zidentyfikowane przez ocenę* Obszary zazwyczaj zagrożone przykurczem i deformacją obejmują zginacze bioder, pasma biodrowo-piszczelowe, ścięgna podkolanowe, zginacze podeszwowe, powięź podeszwową, zginacze łokci, pronatory przedramienia, zginacze i prostowniki długich nadgarstków i palców, mięśnie lędźwiowe i prostowniki szyjne; izolowany przykurcz stawów biodrowych i kolanowych oraz zgięcie podeszwowe, szpotawość tylnej części stopy i przodostopia, zgięcie łokciowe, zgięcie lub wyprost nadgarstka oraz stawy palców; oraz deformacja kręgosłupa i ściany klatki piersiowej, w tym skolioza, nadmierna kifoza lub lordoza oraz zmniejszona ruchomość ściany klatki piersiowej.
- Interwencja ortotyczna, szynowanie, odlewanie, pozycjonowanie i wyposażenie:
- AFO do rozciągania w nocy — mogą być najlepiej tolerowane, jeśli zostaną rozpoczęte profilaktycznie w młodym wieku
- AFO do rozciągania lub pozycjonowania w ciągu dnia w fazach nieambulatoryjnych
- Szyny na nadgarstek lub rękę do rozciągania zginaczy/prostowników palców długich i nadgarstków — zwykle w fazach nieambulatoryjnych
- Odlewanie seryjne – w fazie ambulatoryjnej lub nieruchliwej
- Pasywne/zmotoryzowane urządzenia stojące z podparciem — gdy stanie w dobrym ustawieniu staje się trudne, jeśli przykurcze nie są zbyt silne, aby uniemożliwić ustawienie lub tolerancję
- KAFO z zablokowanymi stawami kolanowymi – opcja dla późnych etapów ambulatoryjnych i nieambulatoryjnych
- Niestandardowe siedzenia w ręcznych i zmotoryzowanych wózkach inwalidzkich (solidne siedzenie, solidne oparcie, prowadnice bioder, boczne podpórki tułowia, przywodziciele i zagłówek)
- Elementy wspomagania pozycjonowania na wózkach zmotoryzowanych (przechylanie, odchylanie, podnoszenie podnóżków, podpórka do stania i regulowana wysokość siedziska)
Ćwiczenia i aktywność
Regularna aktywność lub ćwiczenia submaksymalne, aerobowe (np. pływanie i jazda na rowerze) z pomocą w razie potrzeby, unikanie ekscentrycznych i wysokooporowych ćwiczeń, monitorowanie w celu uniknięcia nadmiernego wysiłku, poszanowanie potrzeby odpoczynku i oszczędzania energii oraz ostrożność w przypadku potencjalnie zmniejszonych ćwiczeń krążeniowo-oddechowych wydolność, a także ryzyko uszkodzenia mięśni, nawet przy prawidłowym funkcjonowaniu klinicznym.
Zapobieganie i leczenie upadków i złamań
- Minimalizacja ryzyka upadku we wszystkich środowiskach
- Fizjoterapeuta wsparcie ortopedów w szybkim zespołowym leczeniu złamań kości długich i zapewnieniu związanej z tym rehabilitacji w celu utrzymania zdolności poruszania się i/lub wspomagania stania
Zarządzanie różnicami w uczeniu się, uwagach i przetwarzaniu sensorycznym
Zarządzanie we współpracy z zespołem, oparte na trosce i ocenie
Technologia wspomagająca i sprzęt adaptacyjny
Planowanie i edukacja z oceną, przepisywaniem, szkoleniem i wspieraniem finansowania
Udział
Uczestnictwo we wszystkich dziedzinach życia wspierane na wszystkich etapach
Zapobieganie i leczenie bólu
Profilaktyka bólu i kompleksowe leczenie w razie potrzeby przez całe życie.
AFO = ortezy kostkowo-stopowe. KAFOs=ortezy kolano-kostka-stopa.
Ocena rehabilitacji
Wielodyscyplinarna ocena rehabilitacji obejmuje pomiary pasywnych zakresów ruchu, rozciągliwości mięśni, postawy i ułożenia, siły, funkcji, jakości życia oraz udziału we wszystkich normalnych czynnościach życia codziennego ( panel 2 ; załącznik ).7,32,33,34,35,49,50,51,52,53,54,55,56,57,58,59
Specjalistyczna ocena funkcjonalna obejmuje analizę wzorców ruchu i standaryzowaną ocenę charakterystyczną dla DMD i innych zaburzeń nerwowo-mięśniowych.32,33,60
North Star Ambulatory Assessment (NSAA) i testy funkcjonalne w czasie są podstawowymi klinicznymi ocenami funkcji w okresie ambulatoryjnym i powinny być wykonywane co 6 miesięcy.32,33,34,35,50,54,55,56,61 Testy NSAA i czasowe testy funkcji mają wysoką wiarygodność i wiarygodność, a także korelację między testami w czasie, minimalne klinicznie istotne różnice i możliwości predykcyjne dotyczące funkcjonalnych zmian motorycznych, które są ważne w monitorowaniu progresji klinicznej i ocenie nowych i pojawiających się terapii.32,33,34,35,55,56,62
Identyfikacja optymalnie reagujących zakresów testowych poprawia możliwości predykcyjne, tak jak w przypadku testu 6-minutowego marszu, w którym zrozumienie interakcji między wiekiem, odległością wyjściową i genetyką może umożliwić lepsze projektowanie badań i informować opiekę kliniczną.35,63,64,65,66
Przewidywanie zmian funkcjonalnych w warunkach klinicznych powinno być dokonywane w kontekście możliwości pacjenta, ze świadomością ograniczeń w ocenie opartej na wysiłku, potencjalnych interakcji z zaburzeniami układu mięśniowo-szkieletowego, takimi jak przykurcz i genetyka.66,67
Testy, które przewidują potencjalne nadchodzące zmiany, mogą być wykorzystywane do kierowania proaktywną opieką, na przykład interwencji na poziomie upośledzenia i przyszłych potrzeb sprzętowych. W szczególności, przed ukończeniem 7 roku życia, mogą wystąpić przyrosty w 6-minutowym teście marszu i testach funkcjonalnych z pomiarem czasu. Po 7 latach wynik testu 6-minutowego marszu wynosi mniej niż 325 m, czas stania ponad 30 sekund, czas wchodzenia po schodach ponad 8 s, czas marszu lub biegu 10 m ponad 10–12 s oraz średnia zlinearyzowana NSAA 34 lub mniej (nieprzetworzony wynik 9) wiązała się z większym funkcjonalnym pogorszeniem poruszania się w ciągu kolejnych 12 miesięcy.35,68
Ocena funkcjonalna obejmuje ocenę czynności życia codziennego oraz zapotrzebowania na sprzęt adaptacyjny lub technologię wspomagającą. Dodatkowo do oceny jakości życia można wykorzystać różne narzędzia.69,70,71,72
Coraz powszechniejsze stosowanie standaryzowanych testów u niemowląt i małych dzieci z DMD pojawia się w odpowiednim czasie ze względu na nowe możliwości wczesnej diagnozy dzięki badaniom przesiewowym noworodków i pojawieniu się terapii, które mogą działać najlepiej, jeśli są stosowane we wczesnym dzieciństwie ( załącznik ). Skala Bayley-III rozwoju niemowląt i skala rozwoju umysłowego Griffitha mierzą tempo rozwoju u dzieci i obie mają zdolność uwydatniania wczesnych opóźnień rozwojowych u dzieci z DMD.49,50,73
NSAA, z rewizją, może być używany do testowania dzieci w wieku 3 lat.51,74
Kinematyka stawu biodrowego podczas chodu jest klinicznie istotną miarą wyników w wieku 4–8 lat.75
Inne miary oceniające funkcję antygrawitacyjną, uważane za eksploracyjne w DMD, obejmują Alberta Infant Motor Scale, Rozszerzoną Funkcjonalną Skalę Motoryczną Hammersmith oraz Gross Motor Function Measure.49,61,76
Ocena i interwencja w zakresie uczenia się, uwagi i przetwarzania sensorycznego powinny rozpocząć się w młodym wieku.77,78
U osób starszych, które nie poruszają się, skala Brooke Upper Extremity Scale, skala Egen Klassifikation oraz zgięcie łokcia i siła chwytu reagują na zmiany w ciągu 1–2 lat.57,79,80
z testowaniem teraz obejmującym osiągalny obszar roboczy52,81
oraz ocena funkcji kończyny górnej (wykonanie testu kończyny górnej).51,53,58
Konsekwentne stosowanie tych samych miar funkcjonalnych przez poszczególne kliniki jest zalecane do śledzenia zmian w czasie, z uwzględnieniem, w stosownych przypadkach, nowych ocen. Zaleca się ocenę przez specjalistów rehabilitacji przynajmniej co 4–6 miesięcy przez całe życie, przy czym częstsza ocena jest spowodowana problemem klinicznym, zmianą stanu lub określonymi potrzebami.
Interwencje
Bezpośrednia terapia fizyczna, zajęciowa oraz logopedyczna powinna być prowadzona w warunkach ambulatoryjnych i szkolnych oraz być kontynuowana przez całe dorosłe życie, wzbogacona o terapie zapewniane podczas hospitalizacji oraz w domu ( panel 2 ; załącznik ).
Celem rozciągliwości mięśni i zarządzania ruchomością stawów jest zapobieganie lub minimalizowanie przykurczów i deformacji ( panel 2 ). Niemożność poruszania stawem w pełnym zakresie ruchu, przewlekłe statyczne pozycjonowanie, nierównowaga mięśni wokół stawu i zmiany zwłóknieniowe w mięśniach powodują zmniejszoną rozciągliwość mięśni i przykurcze stawów.7
Ograniczone wzorce oddychania i zwłóknienie mięśni międzyżebrowych zmniejszają ruchomość ściany klatki piersiowej. Utrzymanie pasywnych zakresów ruchu, rozciągliwości mięśni, ruchomości ściany klatki piersiowej i symetrii może zoptymalizować ruch i funkcjonalne pozycjonowanie, utrzymać poruszanie się, zapobiegać stałym przykurczom i deformacjom, zoptymalizować czynność oddechową i zachować integralność skóry.7
Zarządzanie mięśniowo-szkieletowe wymaga podejścia zespołowego, z udziałem specjalistów nerwowo-mięśniowych, fizjoterapeutów, terapeutów zajęciowych, lekarzy rehabilitacji, ortopedów i chirurgów ortopedów.
Zapobieganie przykurczom i deformacjom wymaga codziennego biernego rozciągania stawów, mięśni i tkanek miękkich zagrożonych uciskiem; wspomaganie ruchu poprzez zmniejszenie wpływu grawitacji i optymalizację biomechaniki w celu umożliwienia bardziej aktywnego ruchu; techniki terapii manualnej i przedłużonego wydłużania tkanek miękkich; oraz optymalne pozycjonowanie, w tym zindywidualizowane stosowanie szyn, interwencje ortopedyczne, urządzenia stojące, odlewanie szeregowe oraz niestandardowe elementy do siedzenia i pozycjonowania mocy w urządzeniach do poruszania się.7,82
Codzienny profilaktyczny program rozciągania w domu83
należy rozpocząć przed utratą biernych zakresów ruchu pod kierunkiem fizjoterapeutów i terapeutów zajęciowych. Rozciąganie jest zalecane w obszarach, o których wiadomo, że są zagrożone przykurczem lub deformacją ( panel 2 ). Regularne rozciąganie kostki, kolana i biodra powinno rozpocząć się wkrótce po postawieniu diagnozy i być kontynuowane w wieku dorosłym. Rozciąganie kończyn górnych jest szczególnie ważne po utracie możliwości poruszania się.7
Dodatek zawiera przegląd rozważań dotyczących pielęgnacji różnych urządzeń wspomagających i ułatwiających poruszanie się, w tym ortez stawu skokowo-stopowego, ortez stawu skokowo-stopowego, odlewania seryjnego, urządzeń stojących oraz ręcznych i zmotoryzowanych urządzeń do poruszania się.7
Wózki inwalidzkie z napędem elektrycznym typu stand-and-drive są obecnie często używane zamiast ortez stawu kolanowego i stopy, aby wspierać mobilność w pozycji stojącej. Takie ortezy mogą nadal być odpowiednim wyborem w niektórych sytuacjach, ale powinny być postrzegane jako narzędzia terapeutyczne, a nie funkcjonalne, uzupełniające, a nie zastępujące ruchomość zmotoryzowaną.84,85
Ponadto innowacje technologiczne — od prostych urządzeń (np. podnoszone tace na kolana i adaptacyjne słomki) do bardziej zaawansowanych technologii (np. robotyka, funkcje Bluetooth umożliwiające zdalną aktywację urządzeń, sterowanie środowiskowe na podczerwień, smartfony, tablety, komputery i zaawansowany dostęp funkcje, takie jak aktywacja głosowa w domu) — można zoptymalizować funkcję. Możliwe wyposażenie adaptacyjne i remonty domów obejmują podnośniki dla pacjentów (podnośniki) do bezpiecznego przenoszenia, rampy, podnośniki schodowe, wyposażenie łazienkowe lub remontowe, specjalne łóżka i materace oraz modyfikacje pojazdów. Opiekunowie opieki osobistej mogą pomóc zoptymalizować niezależność i uczestnictwo.
Fizjoterapeuci przepisują, monitorują i ukierunkowują ćwiczenia, które mogą zapobiec niepotrzebnemu siedzącemu lub nieruchomemu trybowi życia i związanym z tym problemom izolacji społecznej i nadwadze. Jednak wpływ ćwiczeń na degenerację mięśni w dystrofinopatiach, chociaż nie do końca poznany, może obejmować uszkodzenia spowodowane kruchością strukturalną mięśni, nieprawidłowościami metabolicznymi, nieprawidłowościami tlenku azotu przyczyniającymi się do niedokrwienia podczas wysiłku oraz zmniejszoną zdolnością wysiłkową.7,86,87,88,89
Należy unikać ekscentrycznej aktywności mięśni lub ćwiczeń fizycznych oraz ćwiczeń oporowych lub treningu siłowego.7,90
Zalecane są ćwiczenia lub aktywność aerobowa o mniejszej wartości, szczególnie na początku choroby – unikanie nadmiernego wysiłku i przepracowania oraz umożliwienie odpowiedniego odpoczynku.7
Pływanie jest wysoce zalecane od wczesnego etapu chodzenia i może być często kontynuowane aż do dorosłości.7
Jazda na rowerze została zalecona jako submaksymalna forma aktywności aerobowej,91,92
a wspomagana jazda na rowerze i ruch wspomagany przez roboty mogą być stosowane w wieku dorosłym. Bezpieczna aktywność fizyczna może być wspierana przez odpowiedni sprzęt adaptacyjny i technologię wspomagającą.
Ból należy oceniać i leczyć u osób w każdym wieku.7,59,93
Interwencje wymagają kompleksowego zarządzania zespołem, w tym fizjoterapii, korekcji postawy, interwencji ortotycznych i szynowania, ulepszeń wózka inwalidzkiego i łóżka, które umożliwiają niezależną zmianę ciężaru, zmianę pozycji i zmniejszenie nacisku oraz podejścia farmakologiczne. Ból pleców, szczególnie w kontekście leczenia glikokortykosteroidami, powinien skłaniać do oceny złamań kręgów.7
Szczegółowe omówienie profilaktyki i leczenia złamań przedstawiono w części 2 niniejszego Przeglądu.
Postępowanie endokrynologiczne
Powikłania endokrynologiczne DMD i jej leczenia obejmują upośledzenie wzrostu, opóźnione dojrzewanie i niedoczynność kory nadnerczy. Cele opieki endokrynologicznej to monitorowanie wzrostu i rozwoju, identyfikacja i diagnozowanie niedoborów hormonalnych, zapewnienie hormonalnej terapii zastępczej, gdy jest to wskazane, oraz zapobieganie zagrażającemu życiu przełomowi nadnerczowemu. Opublikowano kilka istotnych ekspertyz i recenzji,94,95,96 ale dane są skąpe na temat bezpieczeństwa i skuteczności terapii hormonem wzrostu i testosteronem u osób z DMD. Poniższe rozważania dotyczące opieki są oparte na dowodach i doświadczeniach wynikających ze stosowania tych terapii w innych chorobach, z modyfikacjami do stosowania w DMD (Rysunek 4).

Rysunek 4:
Oceny i interwencje dotyczące upośledzonego wzrostu i opóźnionego dojrzewania płciowego u pacjentów z dystrofią mięśniową Duchenne’a
Wzrost
Zaburzenie wzrostu liniowego jest powszechne u osób z DMD i nasila je leczenie glikokortykosteroidami.39,97
Wzrost liniowy należy oceniać co 6 miesięcy aż do zakończenia dojrzewania i osiągnięcia ostatecznego wzrostu. Wysokość stania jest najwłaściwszą miarą u osób chodzących. Wysokość należy wykreślić i śledzić na znormalizowanej krzywej wzrostu. Ponadto regularną ocenę wzrostu za pomocą nie stojącego miernika wzrostu należy rozpocząć na etapie chodzenia, aby umożliwić dokładniejszą ocenę po utracie możliwości poruszania się. Rozpiętość ramion, długość kości łokciowej, długość kości piszczelowej, wysokość kolan i mierzona odcinkowo długość w pozycji leżącej zostały wykorzystane do oceny wzrostu u dzieci nie poruszających się;98
jednak żadna z nich nie została zwalidowana w populacji DMD i wszystkie wymagają specjalistycznego szkolenia lub specjalistycznego sprzętu. Sugerujemy, aby każda instytucja wybrała i stosowała środek, który najlepiej sprawdza się w jej konkretnym środowisku klinicznym.
Spadek trajektorii wzrostu, o czym świadczy przekroczenie w dół percentyla wzrostu lub roczna prędkość wzrostu mniejsza niż 4 cm na rok, jest zgodne z upośledzonym wzrostem liniowym i wskazuje na potrzebę skierowania do endokrynologa. Osoby o wzroście poniżej trzeciego percentyla powinny być kierowane, niezależnie od trajektorii wzrostu. Ocena upośledzonego wzrostu liniowego powinna obejmować standardowe testy przesiewowe w celu oceny hormonów wydzielania wewnętrznego lub innych nieprawidłowości związanych z zaburzeniami wzrostu. Niewiele danych wskazuje na bezpieczeństwo i skuteczność rekombinowanego ludzkiego hormonu wzrostu w populacji DMD. Jedno retrospektywne badanie wykazało krótkoterminową korzyść w zakresie prędkości wzrostu; jednak niektórzy chłopcy z DMD mieli skutki uboczne, takie jak nadciśnienie śródczaszkowe, nietolerancja glukozy i progresja skoliozy.99
Żadne z opublikowanych badań dotyczących rekombinowanego ludzkiego hormonu wzrostu nie prowadziło obserwacji pacjentów do ich ostatecznego wzrostu i żadne badania nie były wystarczająco duże, aby wiarygodnie ustalić, czy terapia rekombinowanym ludzkim hormonem wzrostu ma negatywny wpływ na czynność mięśni lub inne działania niepożądane. Ponadto pojawiły się teoretyczne obawy, że wysoki wzrost może być szkodliwy dla funkcji mięśni w DMD.100,101
Dopóki nie będzie dostępnych więcej dowodów, nie zaleca się rutynowego stosowania rekombinowanego ludzkiego hormonu wzrostu w leczeniu zaburzeń wzrostu związanych z DMD. Zamiast tego decyzja o leczeniu rekombinowanym ludzkim hormonem wzrostu powinna opierać się na dokładnym omówieniu potencjalnych zagrożeń i korzyści terapii, najlepiej zarezerwowana dla osób z nieprawidłowymi wynikami testów stymulacji hormonu wzrostu.
Dojrzewanie
Opóźnione dojrzewanie płciowe spowodowane hipogonadyzmem jest potencjalnym powikłaniem terapii glikokortykosteroidami i może powodować stres psychiczny i pogarszać jakość życia. Brak dojrzewania do 14 roku życia wymaga szybkiego skierowania do endokrynologa. W celu potwierdzenia rozpoznania hipogonadyzmu u osób z oznakami opóźnionego dojrzewania płciowego należy przeprowadzić testy biochemiczne przy użyciu odpowiednich testów pediatrycznych lub ultraczułych. Należy również rozważyć wykonanie zdjęcia rentgenowskiego lewej ręki w celu ustalenia wieku kostnego.
Terapia zastępcza testosteronem jest zalecana w leczeniu potwierdzonego hipogonadyzmu u pacjentów w wieku powyżej 14 lat i może być rozważana u chłopców w wieku powyżej 12 lat leczonych glikokortykosteroidami bez rozwoju pokwitania. Chociaż żadne badania kliniczne nie oceniały konkretnie stosowania testosteronu u chłopców z DMD, jest on uważany za standard postępowania w leczeniu patologicznego opóźnienia dojrzewania płciowego w populacji pediatrycznej i jest zalecany w leczeniu hipogonadyzmu wywołanego glikokortykosteroidami u dorosłych mężczyzn.102
Potencjalne korzyści testosteronu dla zdrowia emocjonalnego i fizycznego zwykle przeważają nad potencjalnymi skutkami ubocznymi, takimi jak zmiany behawioralne, trądzik, zapach ciała, gwałtowny wzrost i zamknięcie nasad kości. Niedawny przegląd retrospektywny wykazał, że testosteron był ogólnie dobrze tolerowany i postrzegany jako korzystny przez osoby z DMD i ich rodziny.103
Próbując naśladować normalny rozwój dojrzewania, substytucję testosteronu należy rozpoczynać od małej dawki i powoli zwiększać do dawek zastępczych dla dorosłych przez kilka lat. Można stosować preparaty domięśniowe lub miejscowe. Stężenie testosteronu powinno być ściśle monitorowane u wszystkich osób. Należy rozważyć ocenę lipidów, hemoglobiny, hematokrytu i glukozy we krwi u leczonych osób. Negatywny wpływ na stan funkcjonalny pacjenta lub czynność serca powinien skłonić klinicystę do rozważenia przerwania terapii testosteronem lub zmniejszenia dawki.
Niewydolność nadnerczy
Niewydolność nadnerczy spowodowana zahamowaniem osi podwzgórze-przysadka-nadnercza (HPA) jest rzadkim, ale zagrażającym życiu stanem, który może rozwinąć się w przypadku nagłego odstawienia glikokortykosteroidów z powodu choroby lub przerwania leczenia.104
Wszystkie osoby, którym przepisano glikokortykosteroidy, powinny być poinformowane o objawach, objawach i postępowaniu w przełomie nadnerczowym i otrzymać receptę na hydrokortyzon domięśniowy do podawania w nagłych wypadkach w domu (50 mg dla dzieci <2 lat; 100 mg dla dzieci lub dorosłych w wieku ≥2 lat). Dawkowanie stresowe hydrokortyzonu w dawce 50–100 mg/m2 dziennie może być również wymagane w przypadku ciężkiej choroby, poważnego urazu lub zabiegu chirurgicznego u osób przyjmujących prednizon lub deflazakort w dawce powyżej 12 mg/m2 dziennie . Terapia glikokortykosteroidami nie powinna być nagle przerywana, ale raczej zmniejszana w ciągu kilku tygodni lub miesięcy, aby umożliwić powrót do osi HPA.105
Protokół sterydowy PJ Nicholoffa jest odpowiednim podejściem do zmniejszania stężenia glikokortykoidów (steroidów) ( ryc. 3 ).36
Postępowanie żołądkowo-jelitowe i żywieniowe
Powikłania endokrynologiczne DMD i jej leczenia obejmują upośledzenie wzrostu, opóźnione dojrzewanie i niedoczynność kory nadnerczy. Cele opieki endokrynologicznej to monitorowanie wzrostu i rozwoju, identyfikacja i diagnozowanie niedoborów hormonalnych, zapewnienie hormonalnej terapii zastępczej, gdy jest to wskazane, oraz zapobieganie zagrażającemu życiu przełomowi nadnerczowemu. Opublikowano kilka istotnych ekspertyz i recenzji,94,95,96 ale dane są skąpe na temat bezpieczeństwa i skuteczności terapii hormonem wzrostu i testosteronem u osób z DMD. Poniższe rozważania dotyczące opieki są oparte na dowodach i doświadczeniach wynikających ze stosowania tych terapii w innych chorobach, z modyfikacjami do stosowania w DMD (Rysunek 5).

Rysunek 5:
Oceny i interwencje dotyczące postępowania żywieniowego, połykania i przewodu pokarmowego u pacjentów z dystrofią mięśniową Duchenne’a
Ocena i planowanie żywienia
Podczas każdej wizyty w poradni dyplomowany dietetyk powinien ocenić stan odżywienia, śledzić wagę i wzrost oraz stworzyć konkretny plan żywieniowy. Dobry stan odżywienia jest definiowany jako waga dla długości lub wskaźnik masy ciała (BMI) dla wieku, który mieści się między dziesiątym a 85 centylem na standardowych wykresach wzrostu. Jeśli nie można obliczyć BMI, ponieważ nie można zmierzyć wzrostu, należy zastosować percentyle wagi w zależności od wieku. Osoby z DMD mają zmieniony skład ciała, więc stosowanie standardowych wykresów wzrostu nie jest optymalne.
Pacjenci i członkowie ich rodzin powinni praktykować zdrowe, zbilansowane odżywianie zgodnie z zaleceniami aktualnych wytycznych dietetycznych dla Amerykanów.108
Należy podkreślić odpowiednią podaż płynów, aby zapobiec odwodnieniu, co zwiększa ryzyko zaparć i dysfunkcji nerek.109
Panel 3 przedstawia ogólny plan żywieniowy dla osób z DMD.110
Ogólny plan żywieniowy
Ten ogólny plan żywieniowy, który powstał na podstawie zaleceń dla ogólnej zdrowej populacji i nie jest specyficzny dla pacjentów z DMD, zapewnia metody oceny zapotrzebowania na energię, białko, płyny i mikroskładniki odżywcze na podstawie referencyjnej wartości spożycia. Aby zaspokoić codzienne potrzeby żywieniowe organizmu, jednocześnie minimalizując ryzyko wystąpienia chorób przewlekłych, dorośli powinni spożywać 45–65% wszystkich kalorii z węglowodanów, 20–35% z tłuszczów i 10–35% z białka. Dopuszczalne zakresy dla dzieci są podobne do tych dla dorosłych, z tym wyjątkiem, że niemowlęta i młodsze dzieci potrzebują nieco większej zawartości tłuszczu w swojej diecie.110
Ogólne potrzeby kaloryczne
Całkowite zapotrzebowanie kaloryczne jest oparte na całkowitym wydatku energetycznym, który jest równy spoczynkowemu wydatkowi energetycznemu (REE) pomnożonemu przez współczynnik aktywności fizycznej.
Kalorymetria pośrednia zapewnia najdokładniejszy pomiar REE, ale REE można również oszacować u leczonych steroidami chłopców ambulatoryjnych z DMD (w wieku 10–17 lat) za pomocą równania wagi Schofielda (REE [kilokalorie] = [17,7 × waga w kg + 657] × 4,182/1000).111
Ze względu na spadek aktywności fizycznej, który towarzyszy utracie zdolności poruszania się, zapotrzebowanie na kalorie może znacznie się zmniejszyć, a REE może być nawet niższy niż REE przed fazą utraty zdolności poruszania się.
Czynniki aktywności fizycznej chłopców w wieku 3-18 lat to siedzący tryb życia (1,00), mało aktywny (1,13), aktywny (1,26) i bardzo aktywny (1,42).
Obliczona energia lub spożycie kalorii będzie musiało zostać zmniejszone, jeśli początkowa energia lub przepisane kalorie nie skutkują utrzymaniem masy ciała lub utratą masy ciała. Jeśli celem jest zwiększenie masy ciała, obliczona wartość spożycia energii lub kalorii będzie musiała zostać zwiększona.
Białko
Zalecana dieta białkowa różni się dla chłopców i mężczyzn w zależności od wieku: zalecana dieta w wysokości 0,95 g/kg masy ciała na dzień jest zalecana dla dzieci w wieku 4–13 lat; Zaleca się 0,85 g/kg dziennie dla osób w wieku 14-18 lat; a 0,80 g/kg dziennie jest zalecane dla mężczyzn w wieku 19 lat lub starszych.
Płyny
Zalecane spożycie płynów (wszystkie napoje, w tym woda pitna) zależy od wagi lub wieku.
Na podstawie wagi, metoda płynów konserwujących Holliday-Segar112
zaleca 100 ml/kg masy ciała dla dzieci o masie ciała 1–10 kg; 1000 ml + 50 ml na każdy kg powyżej 10 kg dla dzieci o masie ciała 10–20 kg; oraz 1500 ml + 20 ml na każdy kg powyżej 20 kg dla dzieci i dorosłych o masie ciała powyżej 20 kg.
W zależności od wieku, dzienne referencyjne wartości spożycia płynów w diecie wynoszą 1,2 l (około 5 filiżanek) dla chłopców i dziewcząt w wieku 4–8 lat; 1,8 l (około 8 filiżanek) dla chłopców w wieku 9-13 lat; 2,6 l (około 11 filiżanek) dla chłopców w wieku 14-18 lat; i 3,0 l (około 13 filiżanek) dla mężczyzn w wieku 19 lat lub starszych.
Mikroelementy
Zalecana dieta dla wieku113
należy przestrzegać, z wyjątkiem przypadku niedoboru witaminy D, który jest zdefiniowany jako 25-hydroksywitamina D poniżej 30,0 ng/ml. Suplement multiwitaminowy lub mineralny jest konieczny, jeśli spożycie kalorii jest niskie.
Monitorowanie zdrowia kości wymaga corocznej oceny spożycia wapnia w diecie i stężenia 25-hydroksywitaminy D w surowicy. Jeśli spożycie wapnia jest mniejsze niż zalecane dla wieku lub jeśli poziom 25-hydroksywitaminy D w surowicy spada do mniej niż 30 ng/ml, należy zapewnić odpowiednią dietę i suplementację składników odżywczych zgodnie z wytycznymi Instytutu Medycyny.114
Więcej informacji można znaleźć w części 2 niniejszego Przeglądu poświęconej zdrowiu kości i leczeniu osteoporozy.
Zagrożenia żywieniowe specyficzne dla DMD
Osoby z DMD są narażone na nadwagę lub otyłość we wczesnym okresie życia, ze zwiększonym ryzykiem niedożywienia lub niedożywienia, gdy zbliżają się do dorosłości ( załącznik ).115,116
We wczesnym dzieciństwie terapia glikokortykosteroidami zwiększa ryzyko nadwagi lub otyłości z powodu zwiększonego apetytu i spożycia kalorii oraz retencji sodu i płynów. Utrata możliwości poruszania się prowadzi do zmniejszenia aktywności, co zmniejsza zapotrzebowanie kaloryczne i zwiększa ryzyko nadwagi lub otyłości. Aby zaradzić tym zagrożeniom, klinicysta powinien opracować plan żywieniowy zawierający szczegółowe zalecenia dotyczące spożycia kalorii, białka, mikroelementów i płynów (panel 3). Zapotrzebowanie kaloryczne szacuje się, obliczając spoczynkowy wydatek energetyczny i korygując poziom aktywności (panel 3). Zdrowe nawyki żywieniowe, zgodnie z wytycznymi Komitetu ds. Żywienia Amerykańskiej Akademii Pediatrycznej w zakresie zapobiegania otyłości, powinny być przestrzegane przez całą rodzinę (dodatek ).117
Jeśli przyrost masy ciała jest nadmierny, należy opracować plan leczenia otyłości, który uwzględnia zarówno dietę, jak i aktywność fizyczną.
Zaburzenia połykania (dysfagia) są częste i często postępujące u pacjentów z DMD. Ocena wyprzedzająca dysfagii jest ważna i powinna być przeprowadzana regularnie.118
Pytania przesiewowe powinny koncentrować się na odczuwanych trudnościach w połykaniu płynów i pokarmów stałych, postrzeganiu zatykania gardła, czasie niezbędnym do zjedzenia przeciętnego posiłku oraz zakłócaniu jedzenia na jakość życia.119
Jeśli pacjent odpowie na pytania przesiewowe twierdząco, należy skonsultować się z logopedą w celu przeprowadzenia kompleksowej oceny, w tym wideofluoroskopowego badania połykania.120
Osoby często nieumyślnie tracą na wadze przed wystąpieniem klinicznych objawów dysfagii iw ich trakcie. Ich BMI lub percentyle masy ciała mogą spaść z kategorii z nadwagą lub otyłością do normalnego zakresu lub do zakresu niedowagi (niedożywienia) w wyniku trudności w karmieniu i progresji choroby. W załączniku przedstawiono troskę o zmniejszenie ryzyka niedowagi lub niedożywienia w tym okresie przejściowym .
Wczesna i bieżąca dyskusja na temat żywienia przez zgłębnik gastrostomijny może ułatwić szybką interwencję, gdy jest to wskazane klinicznie. Zespół rodzinny i opiekuńczy powinien rozważyć umieszczenie zgłębnika gastrostomijnego jako konieczną i pozytywną interwencję, gdy postępujące osłabienie utrudnia samodzielne odżywianie i połykanie. Wskazania do umieszczenia rurki gastrostomijnej obejmują niedożywienie, które nie odpowiada na interwencje mające na celu poprawę doustnego spożycia kalorii, rozpoznanie umiarkowanej lub ciężkiej dysfagii oraz niemożność utrzymania odpowiedniego nawodnienia. Karmienie zgłębnikiem gastrostomijnym prowadzi do stabilizacji lub poprawy stanu odżywienia u niedożywionych osób z DMD.121
Ocenę korzyści płynących z żywienia przez zgłębnik gastrostomijny należy omówić w kontekście ryzyka związanego z zabiegiem w zakresie układu oddechowego, sercowego i anestezjologicznego.
Częste problemy żołądkowo-jelitowe
Zaparcie jest bardzo częstym powikłaniem DMD.122
Czynniki ryzyka obejmują skrócenie czasu pasażu przez okrężnicę, bezruch, osłabienie mięśni brzucha i odwodnienie (panel 3 ). Może być konieczne codzienne leczenie osmotycznymi środkami przeczyszczającymi, takimi jak glikol polietylenowy, mleko magnezowe lub laktuloza. Lewatywy wsteczne mogą być pomocne w przypadku zaklinowania stolca.
W DMD czynniki ryzyka refluksu żołądkowo-przełykowego obejmują zaburzenia motoryki przełyku, opóźniony czas opróżniania żołądka, terapię glikokortykosteroidami i skoliozę.123
Leczenie refluksu żołądkowo-przełykowego polega na hamowaniu wydzielania soku żołądkowego za pomocą antagonistów receptora histaminowego-2, takich jak ranitydyna, lub inhibitorów pompy protonowej, takich jak lanzoprazol lub omeprazol. Korzyści ze stosowania inhibitorów pompy protonowej muszą być zrównoważone z potencjalnym ryzykiem, w tym częstszym występowaniem pozaszpitalnego zapalenia płuc, przewlekłej choroby nerek i złamań kości.124,125
Podejścia dietetyczne obejmują spożywanie mniejszych, częstszych posiłków i zmniejszenie spożycia tłuszczu w diecie.
W miarę postępu osłabienia mięśni szkieletowych u osób z DMD może wystąpić opóźnienie opróżniania żołądka (gastropareza),123
co może prowadzić do poposiłkowego bólu brzucha, nudności, wymiotów, wczesnej sytości i utraty apetytu. Czas opróżniania żołądka można ocenić za pomocą scyntygraficznego skanu opróżniania żołądka. Opcje leczenia obejmują modyfikację diety, terapię farmakologiczną i żywienie po odźwierniku przez sondę żołądkowo-jelitową.
Wnioski i kierunki na przyszłość
W części 1 tej trzyczęściowej aktualizacji rozważań dotyczących opieki nad DMD przedstawiliśmy wytyczne dotyczące diagnozy oraz postępowania nerwowo-mięśniowego, rehabilitacyjnego, endokrynologicznego i żołądkowo-jelitowego. Najważniejsze z nowych rozważań dotyczących opieki obejmują wytyczne dotyczące opieki nad kobietami nosicielkami DMD; przegląd nowych terapii molekularnych i genetycznych; postępy w ocenie rehabilitacji i pojawienie się bardziej zaawansowanych, technologicznych terapii rehabilitacyjnych; nowe wytyczne dotyczące problemów endokrynologicznych, w tym wzrostu, dojrzewania i niewydolności nadnerczy; oraz nowe spojrzenie na przewidywanie i leczenie powikłań żywieniowych specyficznych dla DMD, takich jak otyłość w połączeniu z terapią glikokortykosteroidami lub utratą możliwości poruszania się oraz niedożywienie w zaawansowanych stadiach DMD.
Możliwość badań przesiewowych noworodków oraz przewidywane pojawienie się genetycznych i molekularnych metod leczenia DMD modyfikujących przebieg choroby oznacza, że w przyszłości coraz ważniejsze będzie wcześniejsze rozpoczęcie leczenia. Jednak optymalny czas na rozpoczęcie nowych terapii będzie kluczowym czynnikiem przy podejmowaniu decyzji o wdrożeniu badań przesiewowych noworodków w kierunku DMD. Nieinwazyjne badania prenatalne w kierunku DMD prawdopodobnie staną się dostępne klinicznie, co pozwoli na wcześniejszą identyfikację dotkniętych chorobą płodów u kobiet bez rodzinnej historii DMD.126
Pojawiają się nowe terapie regeneracyjne dystrofiną, a spodziewane jest ich więcej, a także pojawia się więcej danych na temat najlepszych schematów glikokortykoidów dla pacjentów z DMD.15
Przyszłe rozważania dotyczące opieki będą musiały zająć się rolą nowych związków w ogólnym leczeniu DMD, zwłaszcza w kontekście udowodnionych korzyści z długoterminowej terapii glikokortykosteroidami. Kiedy okaże się, że niektóre z tych nowych terapii są bezpieczne i skuteczne, leczenie DMD można spersonalizować, wybierając najlepszą kombinację terapii dla konkretnej mutacji każdego osobnika. Jeśli chodzi o leczenie hormonalne, konieczne są RCT, aby lepiej zrozumieć ryzyko i korzyści terapii hormonem wzrostu i testosteronem oraz wyjaśnić najlepsze wskazania, czas i schematy dawkowania.
Udoskonalone oceny kliniczne i funkcjonalne zarządzania rehabilitacją są nadal opracowywane, z rozwojem w całym okresie życia. Wraz z postępem technologicznym, nowe terapie będą prawdopodobnie coraz częściej oceniane poprzez monitorowanie aktywności w połączeniu z pomiarami nowych, klinicznie istotnych biomarkerów.127
Robotyka i inne szybkie postępy technologiczne poprawią niezależność, uczestnictwo i jakość życia. Pojawiające się terapie, takie jak terapie regenerujące dystrofinę, mogą poprawić zdolność wysiłkową lub aktywność oraz bezpieczeństwo. Interwencje prowadzone przez fizjoterapeutów, terapeutów zajęciowych, logopedów i ortotystów, wraz z nowymi technologiami, zoptymalizują zarządzanie i funkcjonowanie układu mięśniowo-szkieletowego.128
Wreszcie, w przypadku postępowania żołądkowo-jelitowego i żywieniowego, potrzebne są badania dotyczące spoczynkowego wydatku energetycznego (mierzonego za pomocą kalorymetrii pośredniej) i całkowitego wydatku energetycznego (mierzonego metodą podwójnie znakowanej wody), aby ocenić wydatek energetyczny lub zapotrzebowanie na kilokalorię u osób z DMD. Konkretne strategie żywieniowe, takie jak potencjalna użyteczność diety wzbogaconej w białko lub fruktozę lub suplementacja aminokwasami rozgałęzionymi,129
należy lepiej zrozumieć wpływ stanu odżywienia na wyniki DMD (długość życia, funkcjonowanie i jakość życia). Potrzebne są dalsze badania, aby opracować wykresy wzrostu specyficzne dla DMD, a także dokładne techniki ustalania składu ciała u pacjentów z DMD. Wyjątkowe czynniki warunkujące otyłość u chłopców z DMD należy wykorzystać do opracowania bardziej wykonalnych strategii zapobiegania i leczenia otyłości, w tym opcji farmakologicznych. Opracowanie zaleceń dotyczących bezpiecznej i skutecznej aktywności fizycznej może pozytywnie wpłynąć na stan odżywienia, mobilność i zaangażowanie społeczne przez całe życie pacjentów z DMD.